Tutorial - Functional annnotation¶
This tutorial demonstrates the use of gat for functional annotation. The tutorial follows the analysis in the MacLean et al. (2010) paper introducing GREAT.
The data sets used in this tutorial are available at:
http://www.cgat.org/~andreas/documentation/gat-examples/TutorialFunctionalAnnotation.tar.gz
We are interested if the binding sites of SRF predicted from our ChIP-Seq experiment are enriched in the regulatory domains of genes of specific functions.
The data is in srf.hg19.bed. This bed formatted file
contains 556 high confidence peaks from the analysis of Valouev et al. (2008)
mapped to human chromosome hg19.
Functional annotation with GREAT¶
gat comes with a tool called gat-great that computes
enrichment statistics using the binomial test implemented in GREAT.
To do an analysis as implemented in GREAT, we have prepared a
bed formatted file (regulatory_domains.bed)
with regulatory domains using GREAT’s basal+extension rule.
In GREAT’s definition, the regulatory domain of a gene contains a basal region and an optional extension. The basal region is defined as the region 5kb upstream and 1kb downstream of the transcription start site of a representative transcript. The basal region is then extended up to 1Mb in either direction but only up to the basal region of the closest domains.
In this example, we have used the transcription start sites of the ENSEMBL human protein coding gene set of Ensembl (release 67).
Each gene was replaced with GO terms associated with the gene obtained from the GO Gene Ontology definitions. GO terms were mapped via uniprot identifiers to genes and ancestral ontology terms were inferred.
GREAT excludes assembly gaps in the genome from the analysis. The bed
formatted contigs_ungapped.bed contains all genomic regions
exclusive of assembyl gaps.
We have now all data in place to run a GREAT analysis:
gat-great.py --verbose=5 \
--log=great.log \
--segments=srf.hg19.bed \
--annotations=regulatory_domains.bed \
--workspace=contigs_ungapped.bed \
--ignore-segment-tracks \
--qvalue-method=BH \
--descriptions=go2description.tsv \
>& great.tsv
We also added a file go2description.tsv that contains a table with
descriptions for GO identifiers.
We inserted the table into an SQL database for easy analysis. These are the top 10 results:
| annotation | pvalue | observed | expected | description |
| GO:0015629 | 1.84357379006e-11 | 52 | 18.0074449104 | “actin cytoskeleton” |
| GO:0032796 | 4.22734641877e-09 | 5 | 0.0557976925372 | “uropod organization” |
| GO:0070688 | 1.05832288791e-07 | 7 | 0.358045266782 | “MLL5-L complex” |
| GO:0043229 | 2.56771180099e-07 | 424 | 369.125027893 | “intracellular organelle” |
| GO:0044424 | 3.38999777659e-07 | 457 | 406.64284565 | “intracellular part” |
| GO:0043226 | 3.77332356747e-07 | 424 | 369.977837368 | organelle |
| GO:0072303 | 1.28008163954e-06 | 3 | 0.0198635059219 | “positive regulation of glomerular metanephric mesangial cell proliferation” |
| GO:0001725 | 1.96123184157e-06 | 12 | 2.08962341062 | “stress fiber” |
| GO:0005634 | 2.30813240931e-06 | 295 | 240.687702571 | nucleus |
| GO:0045214 | 2.78146278125e-06 | 11 | 1.79113154372 | “sarcomere organization” |
Validation against GREAT¶
We can check if the results are comparable to the GREAT
server. We submitted our segments to GREAT, downloaded all the results
into srf.great.all.tsv and loaded them into an SQL
database. These are the top 10 results:
| ID | BinomP | ObsRegions | ExpRegions | Desc |
| GO:0015629 | 3.064707e-11 | 51 | 17.68622 | “actin cytoskeleton” |
| GO:0032796 | 4.223825e-09 | 5 | 0.05578831 | “uropod organization” |
| GO:0045214 | 1.057722e-08 | 10 | 0.7800466 | “sarcomere organization” |
| GO:0030863 | 2.205659e-08 | 16 | 2.666392 | “cortical cytoskeleton” |
| GO:0001725 | 1.786459e-07 | 13 | 1.991518 | “stress fiber” |
| GO:0044424 | 2.007378e-07 | 479 | 431.1647 | “intracellular part” |
| GO:0070688 | 2.151319e-07 | 7 | 0.3981916 | “MLL5-L complex” |
| GO:0005634 | 2.556219e-07 | 311 | 251.391 | nucleus |
| GO:0032432 | 2.931657e-07 | 13 | 2.081847 | “actin filament bundle” |
| GO:0043229 | 3.557439e-07 | 443 | 390.9065 | “intracellular organelle” |
The top 10 results are comparable. The same holds generally for fold change values:
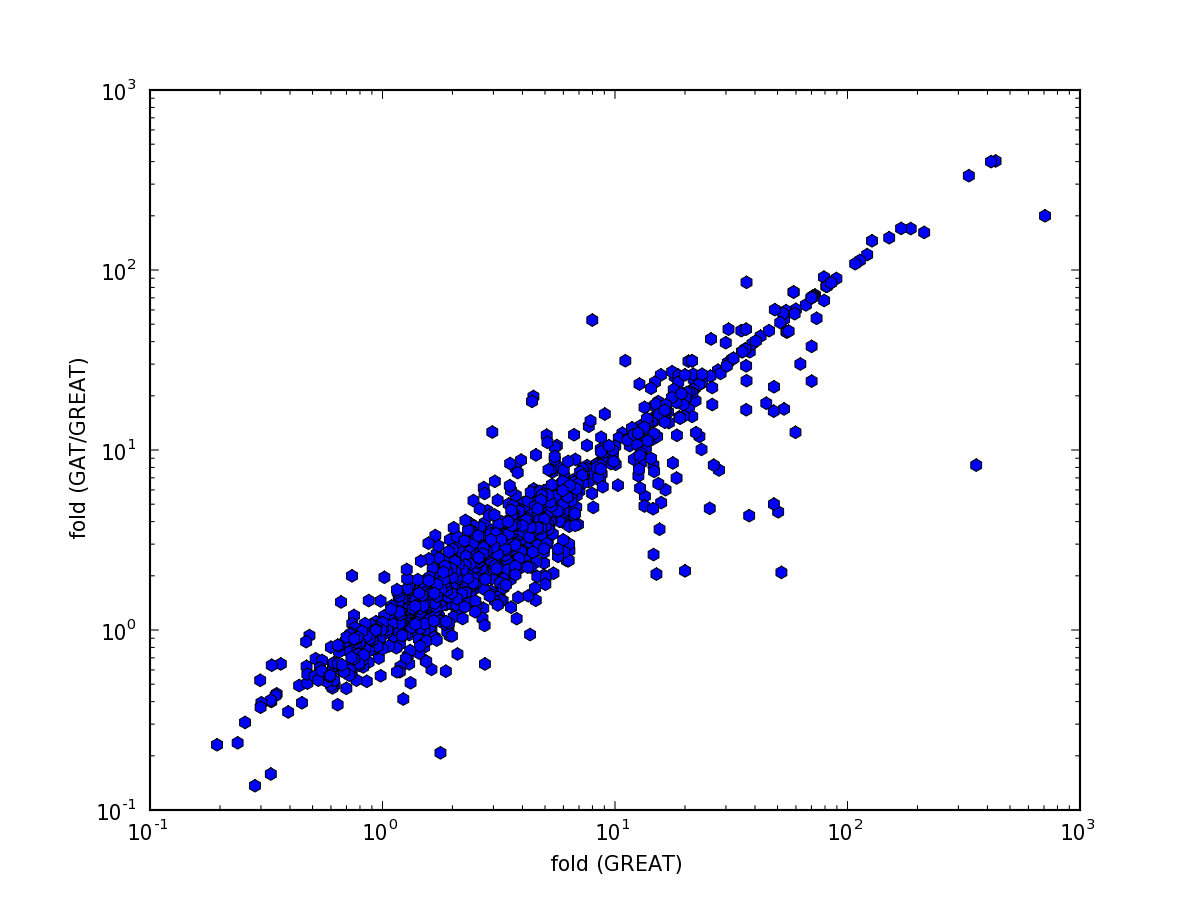
and pvalues:
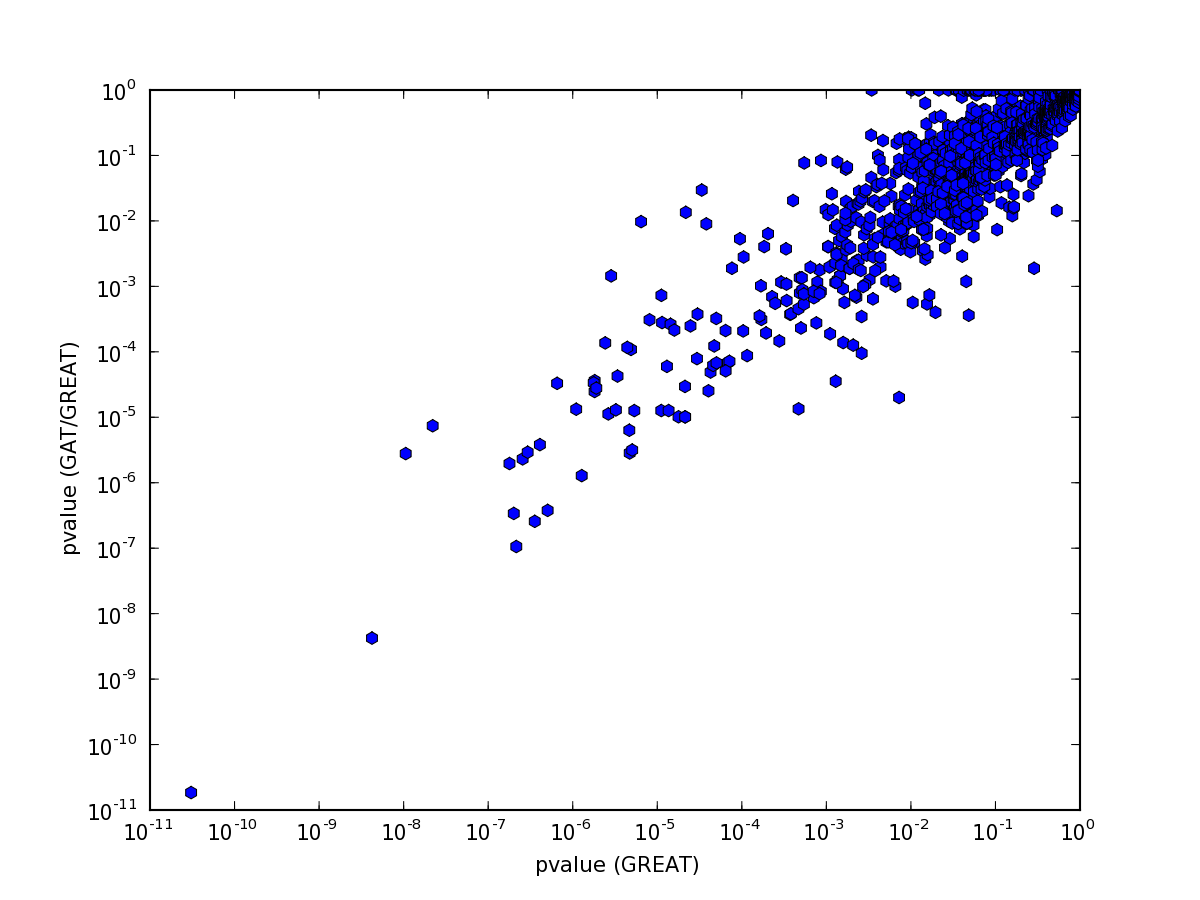
Some differences are to be expected:
- We use the ENSEMBL gene set, while GREAT uses Refseq.
- We use a different definition of representative transcripts.
- Our coordinates of alignment gaps might differ.
- The assignment of GO terms to genes differ.
- The implementations might differ in some details.
Functional annotation with gat¶
Gat can be run with the same input as we used for great:
gat-run.py --verbose=5 \
--log=gatnormed.tsv.log \
--segments=srf.hg19.bed \
--annotations=regulatory_domains.bed \
--workspace=contigs_ungapped.bed \
--ignore-segment-tracks \
--qvalue-method=BH \
--descriptions=go2description.tsv \
--pvalue-method=norm \
>& gatnormed.tsv
Fold changes are very similar:
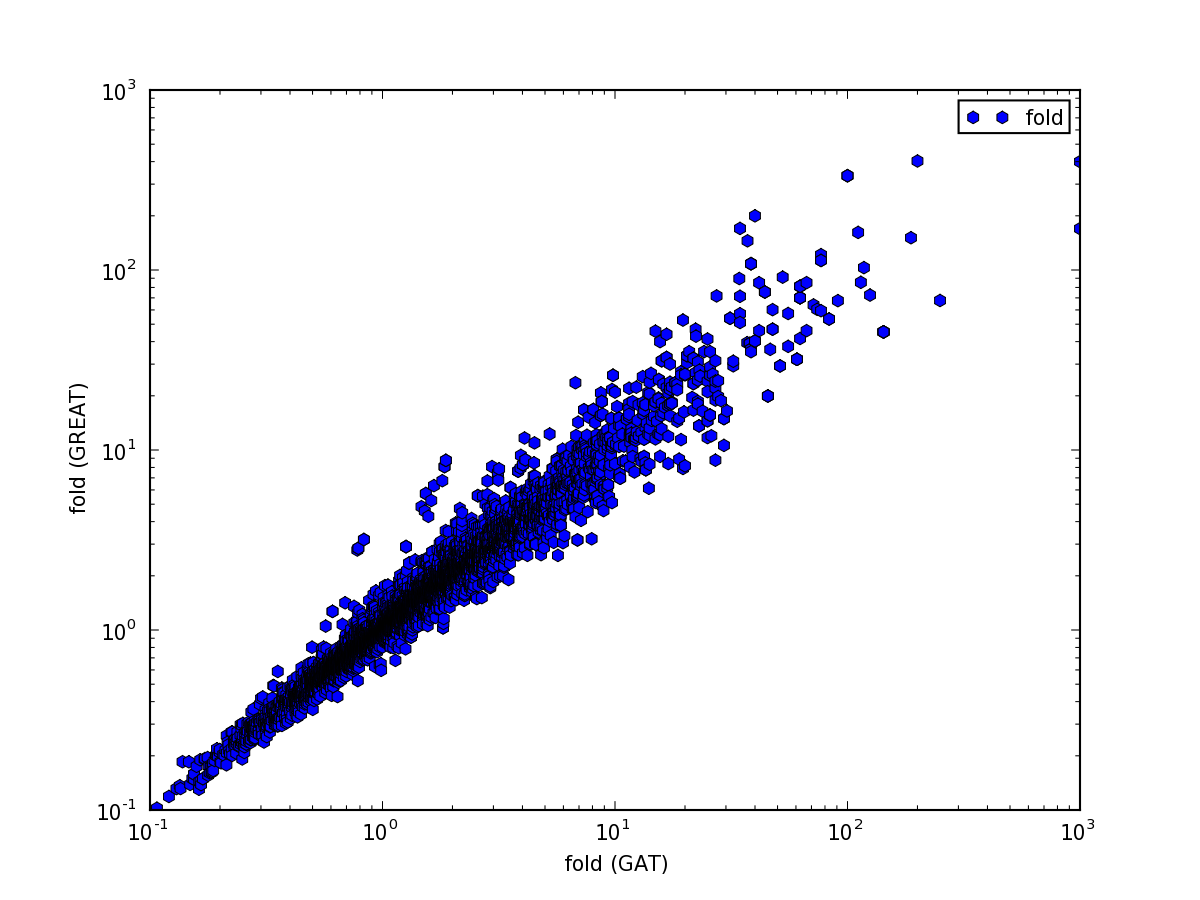
but the p-value comparison shows interesting pattern:
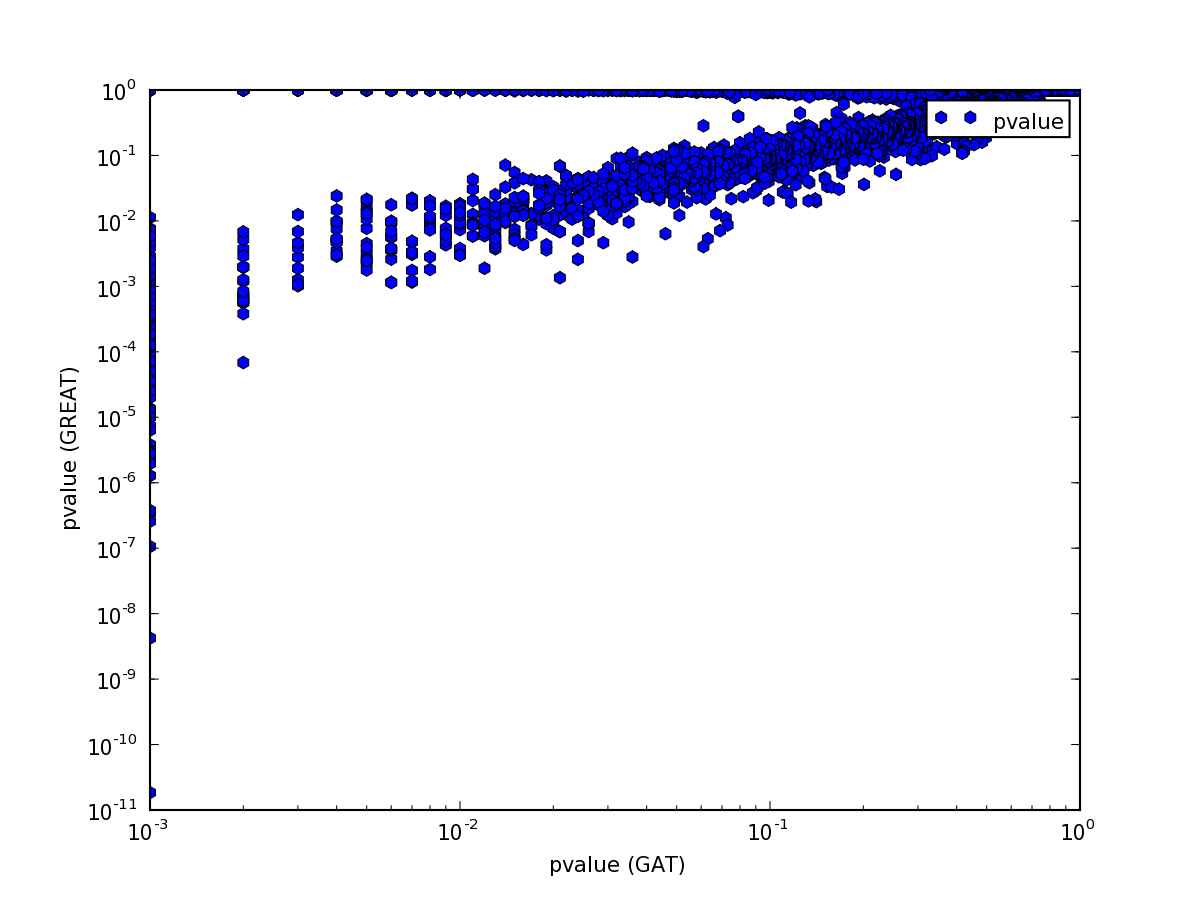
The pattern is explained easily. GREAT computes only the P-Value for enrichment, while GAT computes P-Value both for enrichment and depletion. Indeed, if we only plot p-values for annotations that are enriched, the values are comparable:
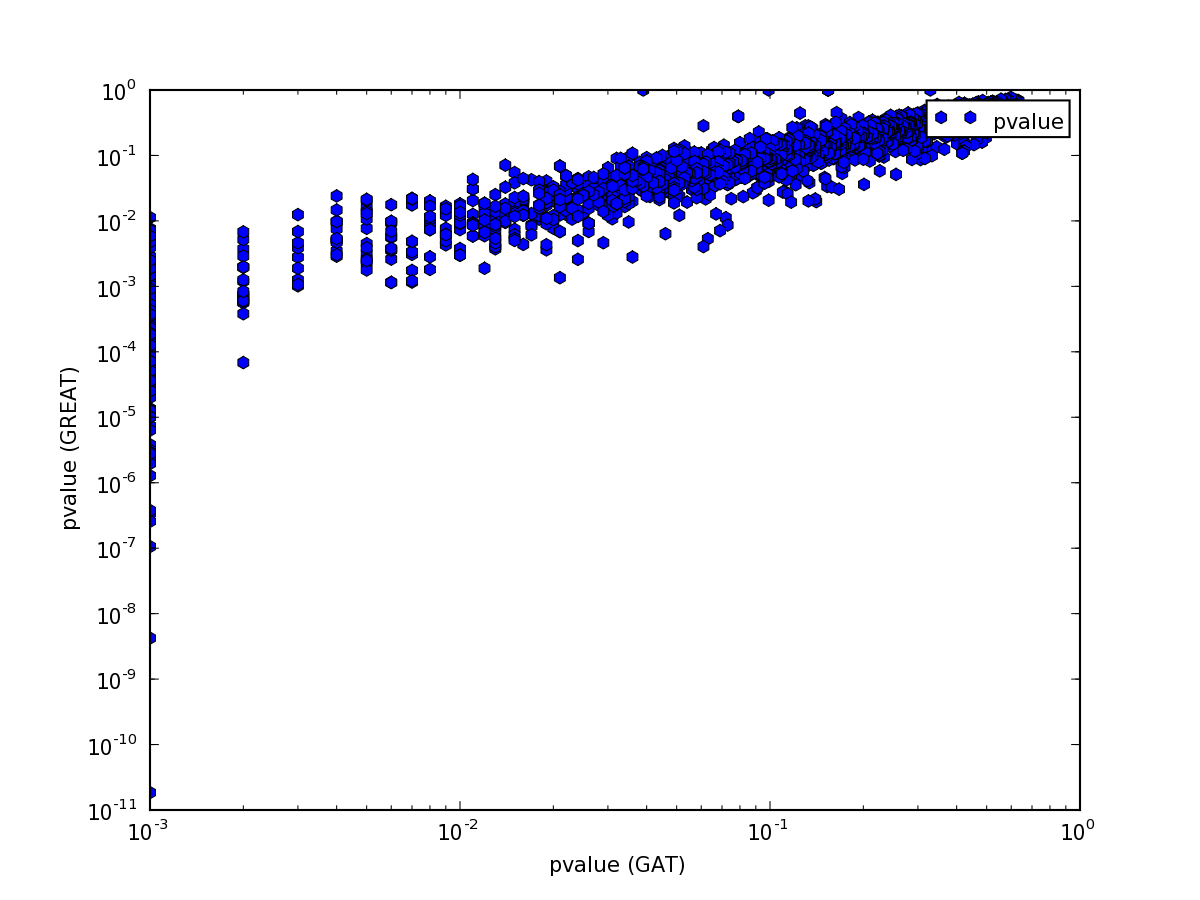
Note how the p-values are very well correlated above 10E-3:
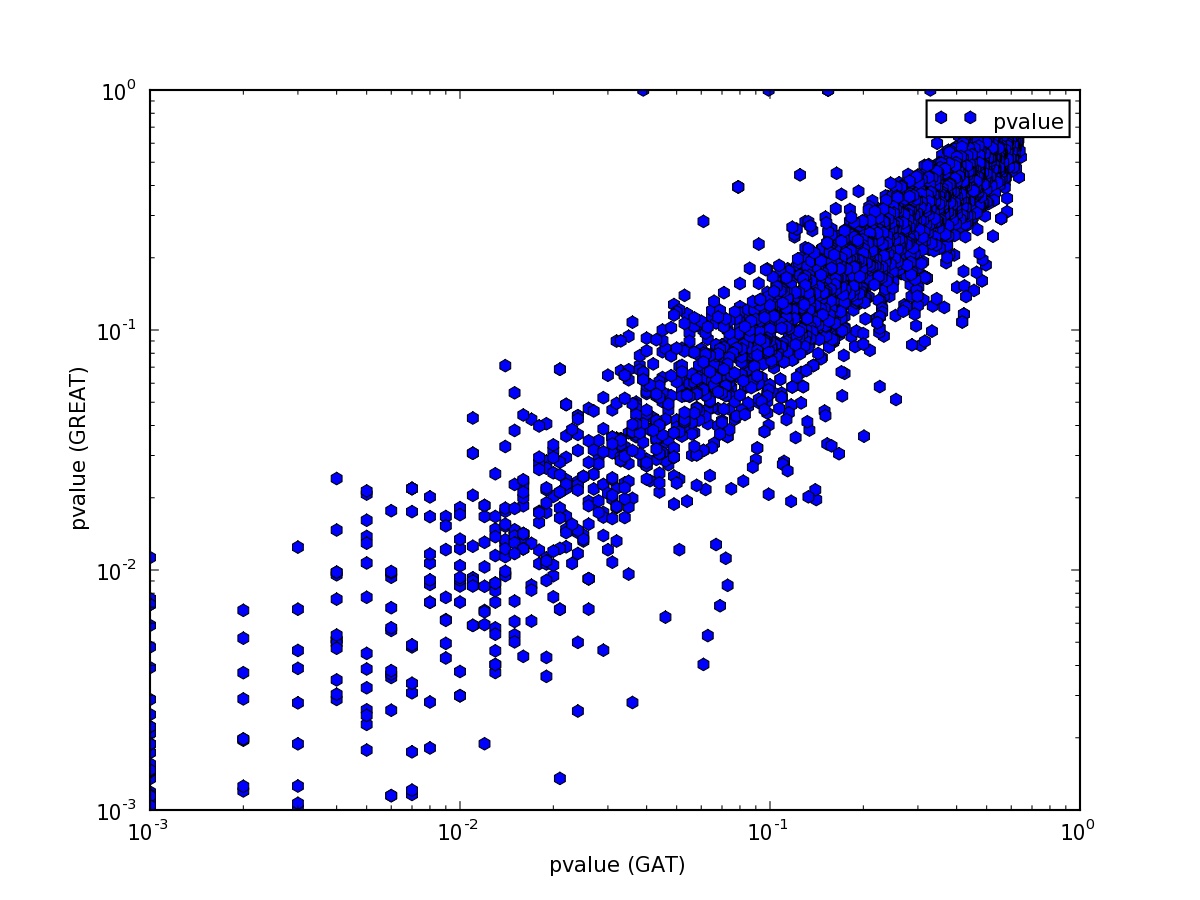
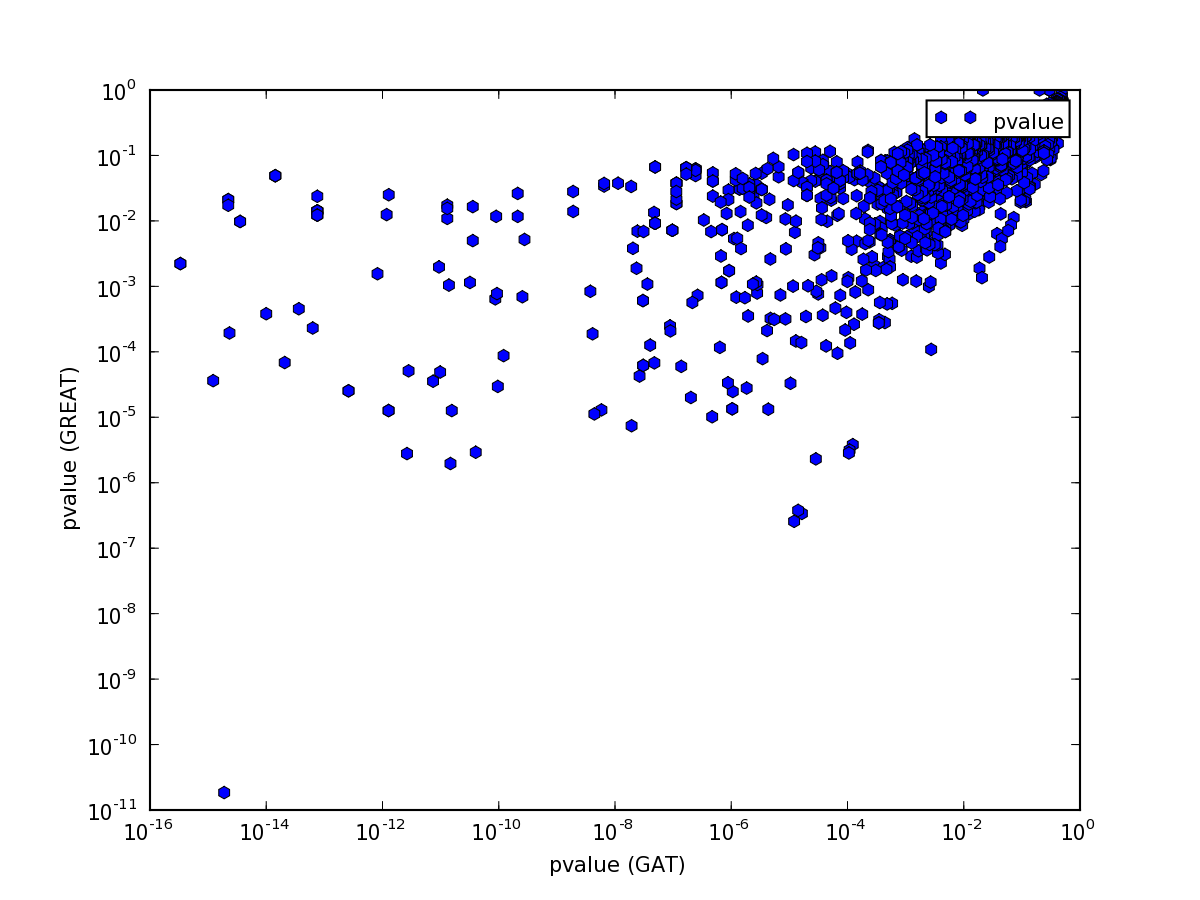
Below a p-Value of 10E-3 the correlation breaks down. Unfortunately,
the lowest empirical p-value is determined by the number of simulations
performed. Adding more simulations will allow us to estimate
lower p-values, but will also increase the runtime. A short-cut is
to extrapolate from lower p-values by adding the option
--pvalue-method=norm:
The table with enriched categories is dominated by small categories with very little expected overlap leading to very large fold changes:
| annotation | pvalue | observed | expected | fold | description |
| GO:0043495 | 0.0 | 200 | 10.8 | 18.5185 | “protein anchor” |
| GO:0070688 | 0.0 | 350 | 16.7 | 20.9581 | “MLL5-L complex” |
| GO:0000212 | 0.0 | 200 | 9.25 | 21.6216 | “meiotic spindle organization” |
| GO:0045896 | 0.0 | 150 | 4.65 | 32.2581 | “regulation of transcription during mitosis” |
| GO:0045897 | 0.0 | 150 | 4.65 | 32.2581 | “positive regulation of transcription during mitosis” |
| GO:0046022 | 0.0 | 150 | 4.65 | 32.2581 | “positive regulation of transcription from RNA polymerase II promoter during mitosis” |
| GO:0046021 | 0.0 | 150 | 4.65 | 32.2581 | “regulation of transcription from RNA polymerase II promoter, mitotic” |
| GO:0071895 | 0.0 | 100 | 2.7 | 37.037 | “odontoblast differentiation” |
| GO:0032796 | 0.0 | 250 | 6.65 | 37.594 | “uropod organization” |
| GO:0021593 | 0.0 | 100 | 2.65 | 37.7358 | “rhombomere morphogenesis” |
For interpretation of the results it is often advisable to remove annotations that are rare.
| annotation | pvalue | observed | expected | fold | description |
| GO:0015629 | 3.3307e-16 | 2600 | 932.458 | 2.7883 | “actin cytoskeleton” |
| GO:0044425 | 5.5445e-11 | 9620 | 13468.369 | 0.7143 | “membrane part” |
| GO:0016021 | 5.7537e-11 | 7870 | 11699.204 | 0.6727 | “integral to membrane” |
| GO:0031224 | 1.2393e-10 | 8220 | 11999.454 | 0.685 | “intrinsic to membrane” |
| GO:0016020 | 5.5517e-08 | 12970 | 16059.768 | 0.8076 | membrane |
| GO:0004888 | 1.0775e-06 | 1000 | 2534.458 | 0.3946 | “transmembrane signaling receptor activity” |
| GO:0030029 | 1.1332e-06 | 2450 | 1278.537 | 1.9163 | “actin filament-based process” |
| GO:0030036 | 1.3077e-06 | 2300 | 1174.087 | 1.959 | “actin cytoskeleton organization” |
| GO:0003779 | 3.2187e-06 | 1900 | 963.501 | 1.972 | “actin binding” |
| GO:0005886 | 5.2155e-06 | 7720 | 10170.117 | 0.7591 | “plasma membrane” |
Currently, we estimate fold enrichment for categories within a workspace that excludes ungapped regions. As before (Tutorial - Interval overlap), a thorough analysis should also exclude regions of low mapability.